My summer student, Kalita, has been digesting oligosaccharides, derivatising them and injecting them into the mass spectrometers in an effort to derive structural information from these complex molecules. We had hoped to use acrylamide gel electrophoresis to visualise the performance of our digests, in the way of Pomin et al (2005).
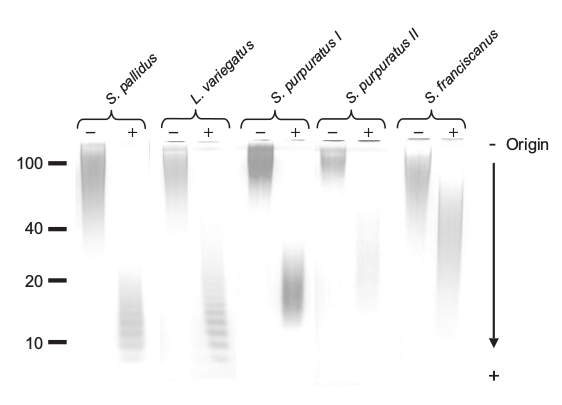
This figure from the paper shows the effect of their hydrolysis technique upon the molecular weight of the oligomer. Note the banding patterns resulting from selective hydrolysis of certain glycosidic bonds. This produces a regular reduction in size of the fragments. We wanted to use this feature to produce polymeric fragments in the <10kDa size rage. These would be amenable to LC-MS/MS, as in Lang et al (2014), allowing us to infer the sequence, functionalisation and bonding of the monomers within the oligomer.
As it turned out our acrylamide gels got lost somewhere amidst The Great Bureaucracy and so, with time running out we cast around for alternate technologies. Enter Yang, et al (2009), who used a similar technique in their paper, but also deployed Size Exclusion Chromatography to illustrate the size-class of fragments produced.
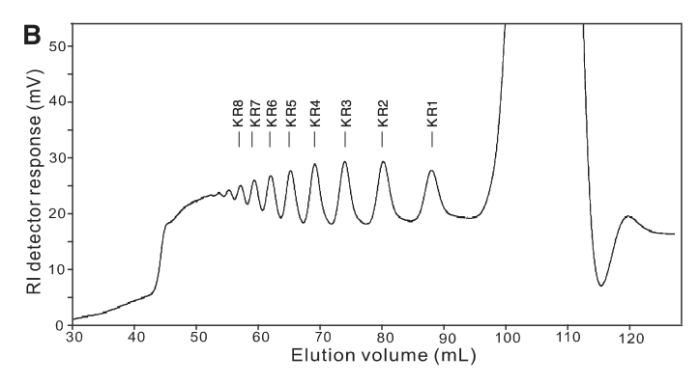
The thing is we didn’t have any GPC or SEC columns. 😦
So we decided to try making our own! 😀
Fortunately or chemical store had a shelf of old bottles of dextran and other GPC or ion-exchange substrates. We dug up a protocol from an MSc thesis by Wilfred Mak in which he’d used an anion exchange substrate to determine the molecular weight of intact sulfated fucan oligosaccharides, rifled through the stores to find some substrates that looked about right and away we went!
We started out with a biuret:

At the bottom, hidden by the blue compression screw, is a plug of deactivated glass wool with a few mL of sand on top of that and then the white dextran gel. This was the first addition of substrate and settling. After topping it up we have a column of about 40cm length. This type of column is purely gravity-fed. You add sample and running buffer at the top and wait for the head of fluid to pass through the column, collecting fractions through the tap at the bottom. This can take hours.
While Kalita was putting this together I was looking at some of the old silica particle LC columns I had and wondering if I might dismantle them, remove the packing and repack them with the dextran to give a real, high-pressure column. This could be plumbed into one of our conventional LC setups, allowing us to push samples through at a faster rate and giving the option of automated sample injection, data and fraction collection. I had something of a brain wave and realised that I had some Swagelok fittings which would allow me to fit a piece of 1/4″ polypropylene air line with pressure-tight caps and LC fittings at either end to fulfil exactly that function. A couple of hours later Kalita and I were the proud parents of monstrous creation on the left!

The white tube held between the two clamps on the left hand retort is the air line packed with hydrated dextran. The line at the top comes from the Shimadzu LC pump on the right, which is pumping Tris buffer through the column to settle the packing material. We can get a flow of 2 mL/min through the column with a back pressure of about 5 bar. Plenty for LC!
For now our creation is parked until we can get round to doing something cool with it on Monday but watch this space to see the outcome. Our intention is to add an autosampler to the front for sample injection, a Refractive Index Detector and maybe even an electrochemical detector on the outflow to detect what came off the column and possibly even a fraction collector for downstream LC-MS/MS analysis of the fractions! Fun!
Our first goal is to validate the SEC function by injecting a range of proteins stained with Bradford Reagent. We can also try some di- and tri-saccharides along with our oligo digests.
References cited
Lang et al (2014). Applications of Mass Spectrometry to Structural Analysis of
Marine Oligosaccharides. Mar. Drugs 2014, 12, 4005-4030
doi:10.3390/md12074005
Pomin et al (2005). Mild acid hydrolysis of sulfated fucans: a selective 2-desulfation reaction and an alternative approach for preparing tailored sulfated oligosaccharides. Glycobiology vol. 15 no. 12 pp. 1376–1385, 2005
doi:10.1093/glycob/cwj030
Yang et al (2009). Mechanism of mild acid hydrolysis of galactan polysaccharides with highly ordered disaccharide repeats leading to a complete series of exclusively odd-numbered oligosaccharides. FEBS Journal 276 (2009) 2125–2137
doi:10.1111/j.1742-4658.2009.06947.x